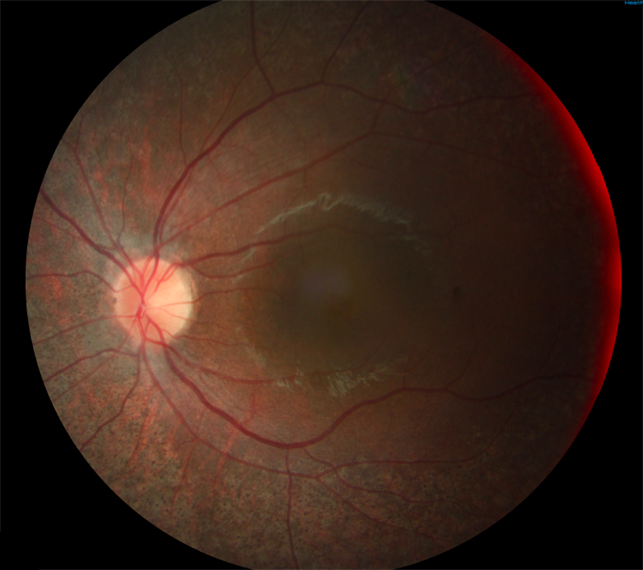
 Синдром Ашера (Ушера, англ. Usher syndrome, врожденная нейросенсорная глухота и пигментный ретинит) представляет собой аутосомно-рецессивное заболевание, включающее в себя зрительные (пигментный ретинит) и слуховые/вестибулярные нарушения. Распространенность синдрома Ашера варьирует от 4 до 17 на 100 000 человек, представлен тремя клиническими подтипами: USH1, USH2 и USH3, которые различаются степенью тяжести нарушений слуха, наличием или отсутствием вестибулярной дисфункции и началом развития пигментного ретинита.
Синдром Ашера (Ушера, англ. Usher syndrome, врожденная нейросенсорная глухота и пигментный ретинит) представляет собой аутосомно-рецессивное заболевание, включающее в себя зрительные (пигментный ретинит) и слуховые/вестибулярные нарушения. Распространенность синдрома Ашера варьирует от 4 до 17 на 100 000 человек, представлен тремя клиническими подтипами: USH1, USH2 и USH3, которые различаются степенью тяжести нарушений слуха, наличием или отсутствием вестибулярной дисфункции и началом развития пигментного ретинита.
Еще в 1858 г. Von Graefe сообщил о 5 случаях пигментного ретинита с тугоухостью с предполагаемой рецессивной формой наследования. Синдром Ушера - редко наблюдаемое заболевание; в общей популяции он встречается с частотой 3 случая на 100 000. Этот синдром диагностируют у 5-8 % больных с наследственной глухотой. Поскольку частота носительства мутантного гена составляет 1 на 100 человек при рецессивном типе наследования, этот синдром манифестирует редко. Точность диагностики возросла с появлением квантитативной аудиометрии. Истинный синдром Ушера характеризуется грубой наследственной сенсорно-нейрональной стабильной (возможно, прогрессирующей) потерей слуха в сочетании с пигментной ретинопатией, не отличимой от пигментного ретинита, которая сопровождается значительным затруднением речи.
Характерными симптомами синдрома Ушера являются ночная слепота и сужение полей зрения, возникающие в возрасте от 5 до 15 лет. Некоторые авторы считают синдром Ушера генетически гетерогенным, другие - фено-типическим проявлением пигментного ретинита. Предполагают, что среди патогенетических механизмов этого заболевания, передаваемого по аутосомно-рецессивному типу, имеется множество трофических нарушений, которые не могут быть определены одним геном. Однако возникновение глазных симптомов связывают с мутацией гена родопсина. Анализ сцепления указывает на локализацию дефекта в длинном плече хромосомы 4 в области 4(q11- q13).
 В настоящее время синдром Ушера - группа генетически гетерогенных заболеваний, наследуемых аутосомно-рецессивно. По клиническим проявлениям выделяют три основных типа заболевания, причем первые два типа включают несколько форм.
В настоящее время синдром Ушера - группа генетически гетерогенных заболеваний, наследуемых аутосомно-рецессивно. По клиническим проявлениям выделяют три основных типа заболевания, причем первые два типа включают несколько форм.
- Первый тип синдрома Ушера представлен 6 формами.
- При анализе сцепления было показано, что первая форма, названная USH1A, локализуется в районе длинного плеча хромосомы 14 (q32.1- q32.3) между локусами D14S78 и D14S250, но ген пока не клонирован.
- Вторая форма, названная USH1B, является наиболее распространенной: ее диагностируют примерно у 75 % от общего количества больных с первым типом заболевания. Эту форму удалось локализовать на длинном плече хромосомы 11q13.5, в районе ДНК-маркера DUS533. В этом же районе был клонирован один из генов миозина типа VILA. Скрининг этого гена на возможные мутации показал их наличие, основная масса которых приводила к преждевременному терминированию синтеза белка.
- Третья форма первого типа - USH1C была картирована в районе короткого плеча хромосомы 11р15.1 и связана с маркером D11S419. В гене-кандидате, названном гармонином, были обнаружены мутации, приводящие к синтезу укороченного белка, в котором отсутствовали его основные домены.
- USH1D удалось картировать в районе длинного плеча хромосомы 10 между локусами D10S529 и D10S573.
- USH1E - сцепление с ДНК-маркерами D21S1905 и D21S1913, расположенными в длинном плече хромосомы 21q21. Генетическое расстояние между этими маркерами составляет около 15 сМ (сантиморган).
- USH1F, с помощью геномного скрининга была картирована на хромосоме 10 в районе локусов D10S199 и D10S596.
- Второй тип синдрома Ушера включает 2 формы.
- Первая из них - USH2A была картирована в районе длинного плеча хромосомы 1q41 между ДНК-маркерами D1S237 и D1S229. Был клонирован ген-кандидат, названный ушерином. Ген USH2A кодирует белок в 1546 аминокислот, который содержит домены эпидермального фактора роста ламинина и фибронектина типа III. Эти домены обнаруживали и в других белковых компонентах базальной пластинки, внеклеточного матрикса и в адгезивных молекулах клеток. В исследовании дана характеристика интрон-экзонной структуры гена, состоящего из 21 экзона. В гене было обнаружено три мутации (239=242insCGTA, R334W, Т1515М) и описано 12 полиморфных вариантов у больных с легкими и тяжелыми формами синдрома Ушера типа II с прогрессирующим течением и сенсорно-нейрональной потерей слуха. При дальнейшем исследовании этого гена выявлено 15 новых мутаций у 57 неродственных пробандов, в том числе обнаружено три новые миссенс-мутации (C319Y, N346H и C419F). Установлено, что 58 независимых аллелей гена USH2A из 114 содержат патологические мутации, из которых наиболее часто (у 16 % больных) наблюдаемая - 2299delG, является мажорной.
- В районе короткого плеча хромосомы 3(р24.2-р23), в области маркеров D3S1578, D3S3647 и D3S3658, была картирована вторая форма второго типа USH2B.
В двух больших семьях с синдромом Ушера типа II с легкой формой пигментного ретинита не выявлено сцепления ни с маркерами 1q41, ни с хромосомой 3q25, т.е. у членов этих семей гены USH2A и USH3 не являлись причиной заболевания. Однако в дальнейшем было установлено, что при фенотипе синдрома Ушера типа II со слабовыраженными офтальмоскопическими проявлениями пигментного ретинита, который не был диагностирован до третьей декады жизни, имелось сцепление с локусом D5S484 на хромосоме 5q. Анализ гаплотипов длинного плеча хромосомы 5q указывает на то, что новый локус расположен между D5S428 и D5S433. К настоящему времени выделено девять таких семей.
В 1996 г. было обследовано 32 семьи с синдромом Ушера типа III (USH3) из географического изолята в Финляндии. Анализ сцепления позволил локализовать заболевание в длинном плече хромосомы 3(q21- q25) между маркерами D3S1299 и D3S3625, расстояние между которыми составляет около 1 сМ (сантиморган).
 По-видимому, существует еще и митохондриальная форма синдрома Ушера. Мутация С12258А в митохондриальном гене MTTS2 явилась причиной развития пигментного ретинита в сочетании с сенсорно-нейрональной формой потери слуха. Работы по картированию генов пигментного ретинита и синдромальных форм продолжаются.
По-видимому, существует еще и митохондриальная форма синдрома Ушера. Мутация С12258А в митохондриальном гене MTTS2 явилась причиной развития пигментного ретинита в сочетании с сенсорно-нейрональной формой потери слуха. Работы по картированию генов пигментного ретинита и синдромальных форм продолжаются.
В семьях, где имеются дети с синдромом Ушера, риск рождения больного ребенка составляет 25 %.
Различают 4 типа синдрома Ушера:
- тип I - пигментный ретинит и тотальная (абсолютная) глухота при отсутствии вестибулярных функций;
- тип II - пигментный ретинит, частичная глухота и интактная вестибулярная функция;
- тип III - пигментный ретинит, полная глухота, вестибулярная атаксия и в отдельных случаях психоз (синдром Халльгрена);
- тип IV - пигментный ретинит, тотальная глухота и задержка умственного развития.
Другая вариация классификации представляется следующим образом:
- грубая ретинопатия, сопровождающаяся функциональными изменениями,
- глубокая врожденная медленно прогрессирующая сенсорно-нейрональная потеря слуха,
- присутствие или отсутствие вестибулярного ответа на тепловой раздражитель,
- статус спиноцеребеллярный и высшей церебральной функции.
В зависимости от возраста появления симптомов ночной слепоты выделяют два типа синдрома Ушера
- Первый тип характеризуется затруднениями при передвижении в условиях слабой освещенности, возникающими в возрасте 5-6 лет,
- при втором типе нарушение ночного зрения возникает в 12-15 лет.
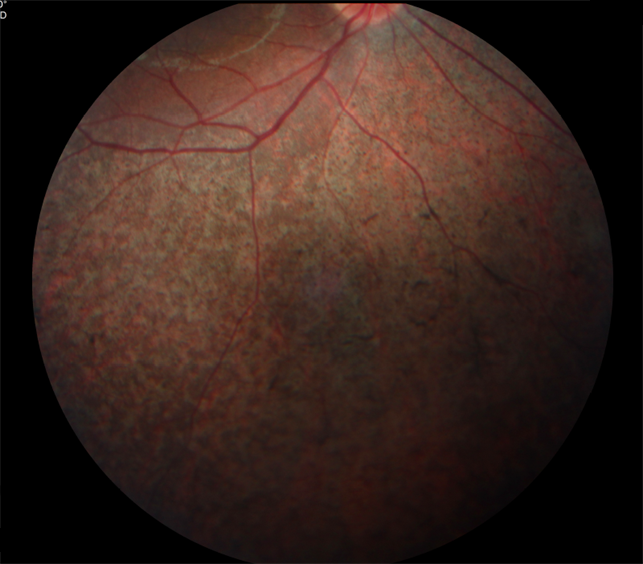
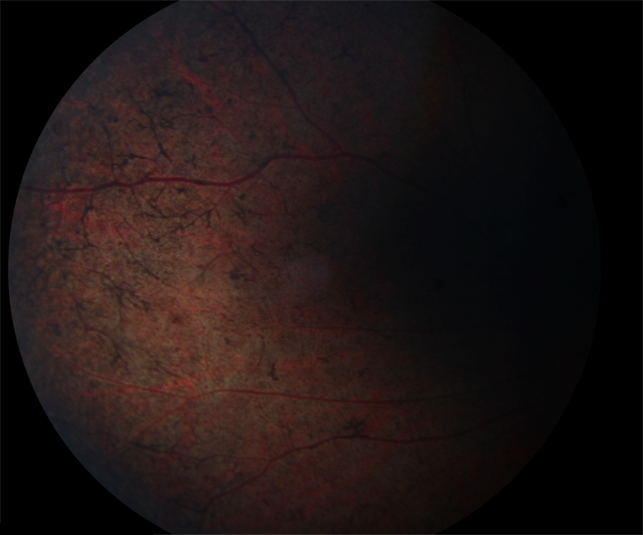
Острота зрения изменяется так же, как при пигментном ретините. При пигментации в макулярной области острота зрения понижается. Прогрессирующий тип заболевания часто наблюдается у больных с синдромом Ушера, которые имеют хорошее зрение. Изменения пигментного эпителия сетчатки схожи с таковыми при макулярной дистрофии типа "бычий глаз" и гипопигментации макулярной области, сочетающейся с пигментным ретинитом.
В последние годы с помощью компьютерной томографии было показано, что у больных с синдромом Ушера с глубокой глухотой имеет место мозжечковая (церебеллярная) атрофия затылочной доли, cavum septum pellucidum, cavum vergae или снижение церебеллярной циркуляции. Картина магнитного резонанса показывает патологически высокий сигнал, свидетельствующий об интенсивности поражения среднего мозга и мозжечка. Однако выявленные изменения мало связаны с клиническими симптомами синдрома Ушера.
В исследованиях, проведенных в последнее десятилетие, установлено нарушение регенерации кинетики зрительного пигмента в фовеа при нормальной световой чувствительности и сохранной остроте зрения.
Лечения синдрома нет. Однако с пациентами должны заниматься социальные работники, нейропсихиатры и воспитатели, используя технику, приспособленную для преподавания по индивидуальным программам. В семьях с близкородственными браками, в которых имеются больные с синдромом Ушера, высок риск рождения других больных детей. Однако определение носителей патологического гена все еще затруднено, хотя в последнее временя совокупность наследственных признаков (сцепление) и достижения молекулярной генетики вселяют надежду.
