- Классификация дистрофий роговицы
- Эпителиальные и субэпителиальные дистрофии
- Эпителиально-стромальные дистрофии, связанные с мутацией гена TGFBI
- Стромальные дистрофии
- Эндотелиальные дистрофии
- Лечение
 Термин «дистрофия» (др.-греч. dystrophe, от dys... — приставка, означающая нарушение + trophe - питание) был впервые использован в медицине в 1884 г. для описания болезней мышц Вильгельмом Эрбом. Уже через 6 лет Артур Греноу применил этот термин для описания характерных поражений роговицы у двух пациентов с зернистой и пятнистой дистрофией. Несколько позже Байбер и Фукс выявляют и другие дистрофические изменения в роговице, предполагая, что патологические процессы являются следствием недостатка питательных веществ, гормонов, кровоснабжения и иннервации. В современной офтальмологии термин «дистрофия роговицы» используется для обозначения группы наследственных заболеваний роговицы, обычно имеющих аутосомно-доминантный тип наследования, двусторонних, симметричных, медленно прогрессирующих, первично поражающих один слой роговицы, не имеющих отношения к внешним воздействиям или системным факторам.
Термин «дистрофия» (др.-греч. dystrophe, от dys... — приставка, означающая нарушение + trophe - питание) был впервые использован в медицине в 1884 г. для описания болезней мышц Вильгельмом Эрбом. Уже через 6 лет Артур Греноу применил этот термин для описания характерных поражений роговицы у двух пациентов с зернистой и пятнистой дистрофией. Несколько позже Байбер и Фукс выявляют и другие дистрофические изменения в роговице, предполагая, что патологические процессы являются следствием недостатка питательных веществ, гормонов, кровоснабжения и иннервации. В современной офтальмологии термин «дистрофия роговицы» используется для обозначения группы наследственных заболеваний роговицы, обычно имеющих аутосомно-доминантный тип наследования, двусторонних, симметричных, медленно прогрессирующих, первично поражающих один слой роговицы, не имеющих отношения к внешним воздействиям или системным факторам.
Дистрофии, как правило, начинаются в молодом возрасте, в 1-2 десятилетие жизни, при этом могут долгое время не иметь клинических проявлений. Дистрофии роговицы можно классифицировать по генетическим нарушениям, гистологическим и биохимическим изменениям, в соответствии с локализацией в анатомически выделяемых слоях роговицы, по степени тяжести течения. Большинство дистрофий роговицы диагностируются по клиническим признакам, таким как сроки манифестации, двустороннее проявление, характерный вид и глубина помутнений, незаинтересованность лимбальной зоны, отсутствие симптомов воспаления. Но необходимо учитывать, что при разных фенотипах одной и той же дистрофии клинические проявления могут широко варьировать. Кроме того, наличие вторичных изменений, таких как рубцы, дегенерация, васкуляризация, может маскировать клинические признаки дистрофии, тем самым создавая диагностические трудности.
Дистрофии роговицы, представляющие собой характерные диффузные помутнения различной формы и локализации, часто сочетающиеся с болевым синдромом за счет рецидивирующих эрозий эпителия, могут быть приняты за вирусную или бактериальную инфильтрацию роговой оболочки глаза, в результате чего назначается неадекватная терапия. Для того чтобы избежать ошибок, необходим осмотр роговицы контрлатерального глаза в прямом и непрямом освещении. Наличие характерного помутнения роговой оболочки контрлатерального глаза может быть признаком ее дистрофии и исключать воспаление. С другой стороны, ряд инфекционно-воспалительных процессов и рубцов могут быть причиной вторичных дегенеративных нарушений, которые клинически напоминают дистрофии роговицы.
Развитие генотипического анализа способствовало значительному прогрессу в изучении дистрофий роговицы. Генетическая характеристика дистрофий роговицы показала их как генетическую, так и фенотипическую гетерогенность, т.е. различные гены могут быть ответственны за один фенотип дистрофии. В то же время один ген (TGFBI) вызывает различные аллельные фенотипы дистрофий.
В 2008 г. была создана и опубликована новая номенклатура каждая позиция по определенной дистрофии представляла собой краткое резюме имеющихся генетических, клинических и морфологических данных. Кроме того, каждой дистрофии была присвоена цифровая категория от 1 до 4 в зависимости от степени доказанности ее существования как отдельной нозологической единицы.
- Категория 1 — это четко определенная дистрофия роговицы, в которой ген был картирован, были определены специфические мутации.
- Категория 2 — это четко определенная дистрофия роговицы, при который были картированы 1 или более определенные хромосомные локусы, но ген(ы) еще предстоит определить.
- Категория 3 — это четко определенная дистрофия роговицы, при которой поражение еще не было картировано в хромосомном локусе.
- Категория 4 предназначается для предполагаемых новых или ранее известных дистрофий роговицы, у которых генная карта не изучена.
Классификация дистрофий роговицы
Эпителиальные и субэпителиальные дистрофии
- Дистрофии базальной мембраны эпителия — К1
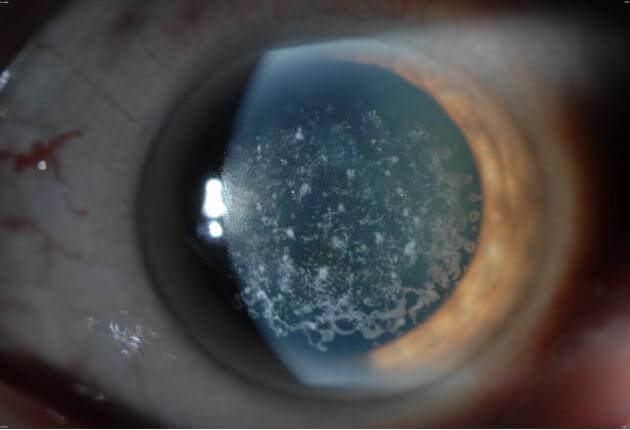
 Дистрофия эпителиальной базальной мембраны (ДЭБМ, дистрофия Когана) — одна из часто встречающихся дистрофий. Хотя она включена в представленную классификацию и поражает один слой роговицы, что типично для дистрофических поражений, только небольшое число случаев имеет доказанное наследование — ген TGFBI (transforming growth factor beta-induced), локус 5q31. В большинстве наблюдений состояние считают специфической реакцией роговицы на внешние воздействия, т.е. дегенерацией, которая обусловлена аномальным восстановлением, созреванием и формированием базальной мембраны, приводящим к образованию патологических комплексов адгезии и ослабленному прикреплению эпителия к строме.
Дистрофия эпителиальной базальной мембраны (ДЭБМ, дистрофия Когана) — одна из часто встречающихся дистрофий. Хотя она включена в представленную классификацию и поражает один слой роговицы, что типично для дистрофических поражений, только небольшое число случаев имеет доказанное наследование — ген TGFBI (transforming growth factor beta-induced), локус 5q31. В большинстве наблюдений состояние считают специфической реакцией роговицы на внешние воздействия, т.е. дегенерацией, которая обусловлена аномальным восстановлением, созреванием и формированием базальной мембраны, приводящим к образованию патологических комплексов адгезии и ослабленному прикреплению эпителия к строме.
Дистрофия встречается у 2% в популяции, чаще у женщин. При биомикроскопии в эпителиальном слое роговицы выявляются сероватые включения, тонкие линии, микрокисты. Эти образования формируют различные типы рисунка в субэпителиальном слое — точечные и микрокистные изменения, географический рисунок, рисунок «отпечатка пальца», «булыжной мостовой». Со временем они могут менять свою локализацию и тип рисунка. Так как эти интраэпителиальные поражения полупрозрачны и могут быть очень маленькими по размеру, биомикроскопия должна проводиться с особой тщательностью с использованием различной техники освещения. При гистологическом исследовании выявляются интраэпителиальные разрастания или многослойные образования базальной мембраны эпителия, которые формируют географический рисунок или рисунок «отпечатка пальца». Точечные изменения представляют собой псевдокисты, содержащие твердые частицы в цитоплазме и скопление клеточного детрита между патологическими разрастаниями мембраны либо в слоях эпителия.
В большинстве случаев дистрофия протекает бессимптомно, но одной из первых жалоб становятся сильные боли по утрам, при открытии глаза. Симптоматика дистрофии тесно связана с синдромом рецидивирующей эрозии роговицы и временным незначительным снижением остроты зрения, что чаще имеет место у пациентов старше 30 лет. . Считается, что от 10 до 30% пациентов с ДЭБМ подвержены рецидивам эрозии роговицы и у 50% больных с рецидивирующей эрозией роговицы выявляются признаки ДЭБМ. При изменениях базальной мембраны в оптическом центре возможен неправильный астигматизм.
- Дистрофия эпителиальной рецидивирующей эрозии — К3.
- Субэпителиальная мукоидная дистрофия — К4.
- Дистрофия роговицы Месманна — К1.
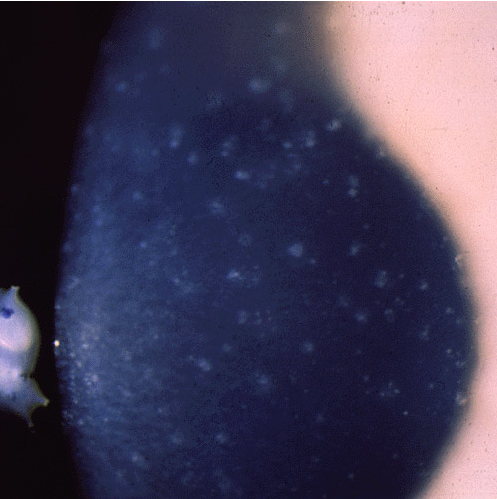 Аутосомно-доминантное заболевание, которое начинается в раннем детском возрасте. Редкая форма дистрофии роговицы, поражающая роговичный эпителий, в котором из-за мутаций генов KRT3 и KRT12, кодирующих кератин, возникают кисты. Происходит образование множественных мелких пузырьков в эпителии роговицы с последующим превращением их в точечные помутнения. По данным электронной микроскопии пузырьки содержат цитоплазматические включения. Роговичная ткань между пузырьками не изменена.
Аутосомно-доминантное заболевание, которое начинается в раннем детском возрасте. Редкая форма дистрофии роговицы, поражающая роговичный эпителий, в котором из-за мутаций генов KRT3 и KRT12, кодирующих кератин, возникают кисты. Происходит образование множественных мелких пузырьков в эпителии роговицы с последующим превращением их в точечные помутнения. По данным электронной микроскопии пузырьки содержат цитоплазматические включения. Роговичная ткань между пузырьками не изменена.
Дефекты эпителия роговицы обусловливают появление болей и ощущение инородного тела. Острота зрения заметно не страдает. Могут отмечаться фотофобия и периодически возникающее затуманивание зрения. Из-за крайне малого размера мутных пятнышек, постепенно образующихся в толще эпителия, болезнь зачастую диагностируют, когда пациент достигает среднего возраста и начинает страдать от фотофобии и снижения зрения. Лечения обычно не требует. В качестве терапии может быть использована фототерапевтическая кератэктомия, но она не ведет к окончательному излечению.
- Эпителиальная дистрофия роговицы Лиша — К2.
Редкая форма дистрофии роговицы. В одном исследовании отмечена ассоциация заболевания с хромосомной областью Xp22.3, но пока не выявлено конкретных генов-кандидатов. Основные признаки дистрофии Лиша – появление полос или хлопьев помутнения на роговице одного или обоих глаз. Иногда помутнения закручиваются водоворотом, следуя типичной картине обновления эпителия роговицы. В цитоплазме эпителиальных клеток из областей помутнения отмечены диффузные вакуоли с неизвестным содержимым. Фототерапевтическая кератэктомия у таких пациентов лишь временно улучшает их самочувствие, через несколько месяцев эпителий обновляется и вновь начинает мутнеть.
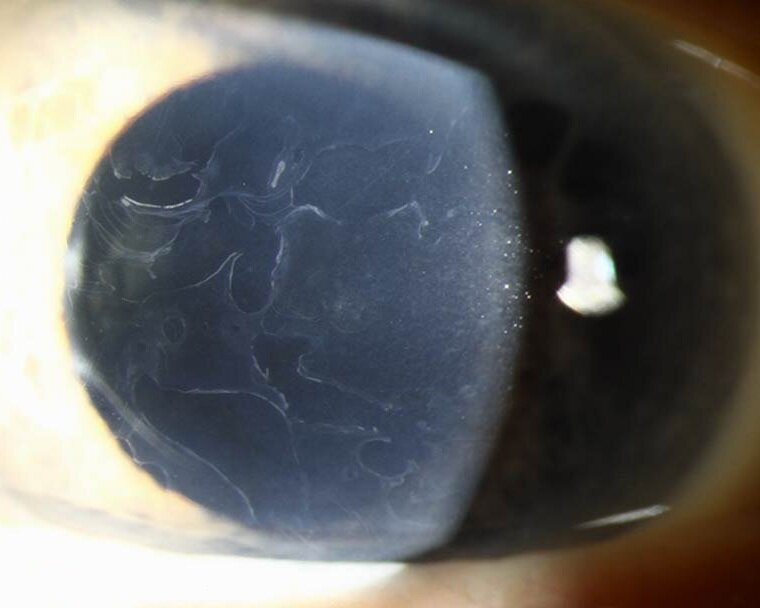
- Студенистая каплевидная дистрофия — К1.
Редкая форма, аутосомно-доминантный тип наследования. Ряд мутаций, вызывающих эту дистрофию, отмечен в гене TACSTD2, однако у некоторых пациентов этот ген не затронут, что говорит о полигенном характере заболевания. При этой дистрофии наиболее заметны гроздеобразные желатинозные массы под эпителием роговицы. В далекозашедшем течении заболевания наблюдается совершенно непрозрачная роговица с множественными каплеобразными помутнениями. В помутневших областях заметны кровеносные каналы. В амилоидных включениях обнаруживается лактоферрин, однако ген лактоферрина не затронут. У пациентов развивается светобоязнь, ощущение постороннего тела в роговице, сильно падает зрение.
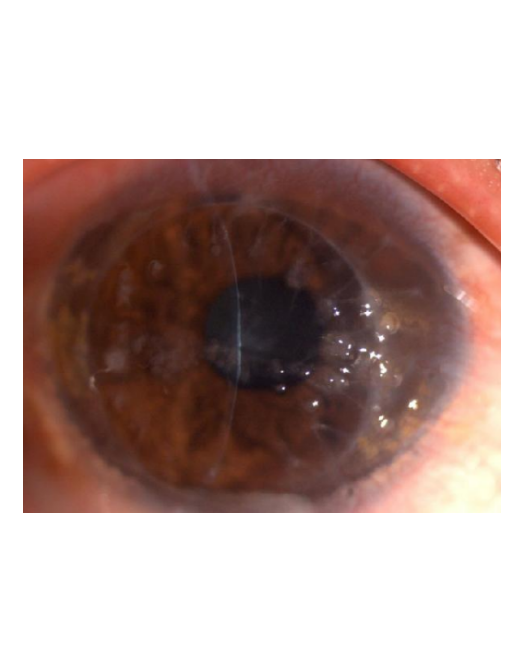
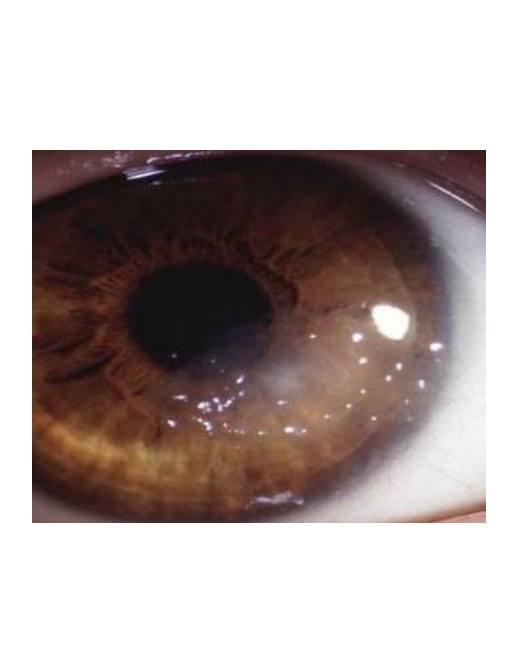
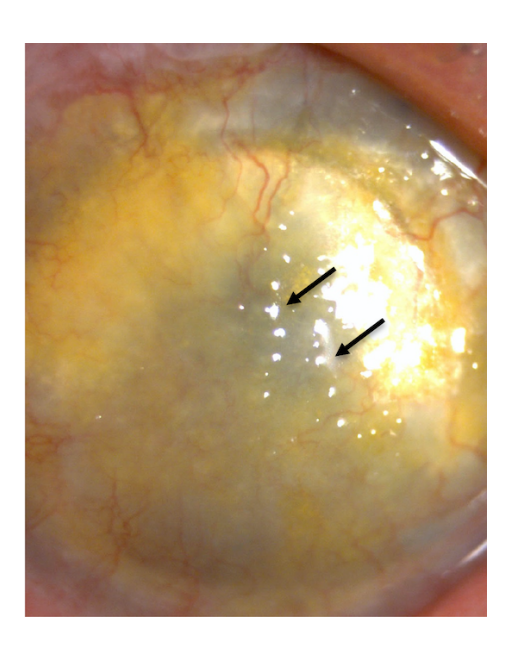
Эпителиально-стромальные дистрофии, связанные с мутацией гена TGFBI
- Дистрофия Рейс—Буклера — К1
Аутосомно-доминантная (ген TGFBI, локус 5q31), билатеральная, центральная дистрофия роговицы с первичным поражением боуменового слоя, начальные признаки которой появляются в первые годы жизни в виде поверхностных линейных и кольцевидных сероватых помутнений, сопровождающихся рецидивирующими эрозиями с выраженным болевым компонентом. К 20-30 годам помутнения прогрессируют, сливаются между собой, сохраняя характерный географический рисунок, распространяясь на среднюю периферию и глубже в строму, развивается диффузное стромальное неинтенсивное помутнение (хейз). Частота эрозий с возрастом заметно уменьшается, ухудшается острота зрения из-за прогрессирования помутнений и неправильного астигматизма. Снижение остроты зрения при этой дистрофии более выражено, чем при сходной дистрофии Тиля-Бенке, из-за большего нарушения регулярности передней поверхности роговой оболочки глаза. При гистологическом исследовании видно, что эпителиальный слой теряет регулярную архитектонику, местами утолщен, повторяет измененный рельеф подлежащей соединительной ткани. Местами отсутствует базальная мембрана эпителия. Боуменов слой замещается листовидной фиброцеллюлярной соединительной тканью с зернистыми отложениями, которая в развитой стадии заболевания распространяется в передние слои стромы.
- Дистрофия роговицы Тиля—Бенке — К1.
Дистрофия боуменовой мембраны II типа. Первоначально считалась проявлением дистрофии роговицы Рейса-Бюклера, но исследование, проведенное в 1995 году М. Кюхлем с коллегами, показало, что две схожие дистрофии являются разными заболеваниями. Некоторые случаи заболевания ассоциированы с хромосомным участком 10q24, другие являются результатом мутации гена TGFBI, кодирующего кератоэпителин. В большинстве случаев пациенты с дистрофией Тиль-Бенке, или дистрофией «медовых сот», рождались со здоровой неизмененной роговицей. Но в первые 10-20 лет жизни начинали развиваться помутнения, выраженные рецидивирующие эрозии, сетчатое рубцевание роговицы и дистрофические изменения в виде «медовых сот» в области боуменовой мембраны.
- Решетчатая дистрофия роговицы, тип 1 — К1.
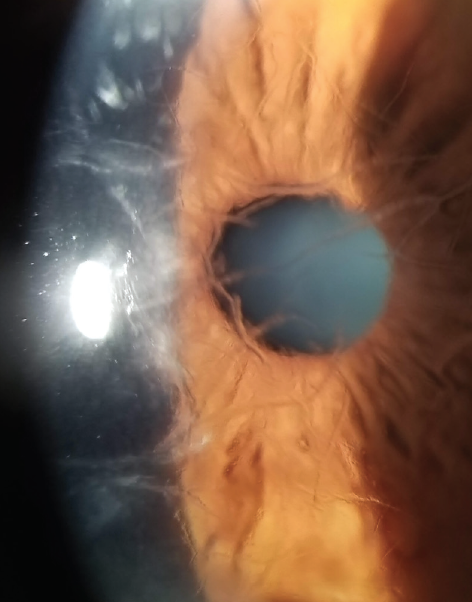 Решетчатая дистрофия роговицы, тип 1, считается классической решетчатой дистрофией роговицы, имеет аутосомно-доминантный тип наследования (ген TGFBI, локус 5q31). При гистологическом исследовании выявляются отложения амилоидного вещества в строме, которые нарушают ламеллярную архитектонику роговицы. Также могут отмечаться эпителиальная атрофия и дегенеративные нарушения базальных эпителиальных клеток, очаговое истончение или отсутствие слоя Боумена.
Решетчатая дистрофия роговицы, тип 1, считается классической решетчатой дистрофией роговицы, имеет аутосомно-доминантный тип наследования (ген TGFBI, локус 5q31). При гистологическом исследовании выявляются отложения амилоидного вещества в строме, которые нарушают ламеллярную архитектонику роговицы. Также могут отмечаться эпителиальная атрофия и дегенеративные нарушения базальных эпителиальных клеток, очаговое истончение или отсутствие слоя Боумена.
При биомикроскопии уже к концу первого десятилетия жизни видны тонкие ветвящиеся, отражающие свет линии и/или субэпителиальные беловатые овальные точки. Линии появляются в центре достаточно поверхностно, распространяясь на периферию и в глубь роговой оболочки глаза, но оставляя периферию, десцеметову мембрану и эндотелий интактными. Диффузное стромальное помутнение матового цвета, как правило, развивается позже, сопровождается рецидивирующими эрозиями. Дискомфорт, боль и зрительные нарушения иногда начинаются уже в первое десятилетие жизни. Зрительные нарушения становятся значительными к четвертому десятилетию, что часто является показанием к кератопластике в этом возрасте.
Решетчатая дистрофия роговицы, тип 2, была внесена в первую редакцию современной классификации лишь условно, исключена из второй, не считается дистрофией роговицы как таковой, а является глазным проявлением семейного амилоидоза. Сопровождается краниальной нейропатией, проявляющейся как лицевой парез, бульбарный паралич, периферической полинейропатией. При биомикроскопии изменения сходны с таковыми при типе 1. Чувствительность роговицы снижена или отсутствует. Острота зрения сохраняется достаточно высокой длительное время, так как дистрофия прогрессирует от периферии к центру роговицы. Развивается симптоматика синдрома сухого глаза. В пожилом возрасте могут возникать рецидивирующие эрозии роговицы. Течение заболевания медленно прогрессирующее. У большинства может не быть серьезных нарушений до седьмого десятилетия. Существует повышенный риск открытоугольной глаукомы.
- Гранулярная (зернистая) дистрофия роговицы, тип 1 — К1.
Имеет аутосомно-доминантный тип наследования (ген TGFBI, локус 5q31), билатеральная, симметричная. Заболевание проявляется в первую декаду жизни. Осмотр за щелевой лампой выявляет хорошо видимые гранулы, которые кажутся белыми при прямом освещении. При ретроиллюминации эти гранулы состоят из чрезвычайно мелких, полупрозрачных точек, выглядят как вакуоли, стеклянные осколки или измельченные крошки. Помутнения не захватывают лимбальную зону. У детей коричневые гранулы, образующие вихревидный рисунок, расположены относительно поверхностно и распространяются к слою Боумена. В дальнейшем гранулы простираются в глубокие слои стромы вплоть до десцеметовой мембраны. Блики и светобоязнь являются относительно ранними симптомами. Так как на начальных этапах регулярность передней поверхности не нарушена, острота зрения остается высокой. С возрастом помутнения прогрессируют, сливаются между собой, острота зрения постепенно снижается. Иногда имеют место рецидивирующие эрозии. В развитых стадиях острота зрения редко снижается менее 0,1. Как правило, к 50-60 годам необходима кератопластика, после которой вероятны рецидивы в течение нескольких лет (от 1 года до 20 лет).
Гранулярная дистрофия роговицы, тип 2 — К1.
Зернистая дистрофия роговицы, тип 2, считается комбинированной — зернисто-решетчатой. Также часто именуется дистрофией роговицы Авеллино по местности, где проживала семья, страдавшая таким заболеванием. Имеет аутосомно-доминантный тип наследования (ген TGFBI, локус 5q31). По данным световой микроскопии помутнения роговицы простираются от базального эпителия до глубокой стромы. Помимо типичных для зернистой дистрофии 1-го типа гиалиновых отложений, в строме присутствует амилоид. Заболевание начинается в первой декаде жизни, но выявляется чаще в подростковом возрасте или в ранней зрелости. Начальными признаками дистрофии, которые выявляются под щелевой лампой, считаются поверхностные стромальные крошечные белесые точки. На следующем этапе между поверхностной и средней стромой появляются кольцевидные или в форме снежинки помутнения. У некоторых пациентов в более глубоких слоях также имеют место решетчатые линии. Как правило, эти линии расположены глубже, чем стромальные помутнения в виде снежинок. На заключительном этапе полупрозрачные плоские крошковидные помутнения, расположенные поверхностно, могут сливаться между собой. У некоторых пациентов проявления дистрофии ограничиваются только множеством белых точек. Пациенты с зернистой дистрофией 2-го типа имеют меньше помутнений, чем лица с зернистой дистрофией 1-го типа. Острота зрения уменьшается с возрастом, так как постепенно вовлекается центральная зона. При эрозиях, которые, как правило, не имеют тяжелого течения, возникают болевые ощущения. Заболевание прогрессирует медленно, несколько быстрее при гомозиготной форме.
Стромальные дистрофии
Для стромальных и ряда эпителиально-стромальных дистрофий характерным является отложение патологических веществ между коллагеновыми фибриллами или в кератоцитах. Это могут быть как нормальные метаболиты, представленные в избыточном количестве, как гликозамингликаны при макулярной дистрофии, так и вещества, не встречающиеся в роговой оболочке в норме (амилоид, холестерол, гиалин).
- Пятнистая дистрофия роговицы — К1.
Макулярная (пятнистая) дистрофия роговицы. Имеет аутосомно-рецессивный тип наследования, ген CHST6 (сarbohydrate sulfotransferase 6 gene), локус 16q22. Морфологические изменения связаны с избыточным отложением в строме, эндотелии и десцеметовой мембране внутриклеточно и внеклеточно гликозаминогликанов. Заболевание проявляется в детском возрасте диффузным помутнением стромы, распространяющимся до лимба; позже обнаруживаются поверхностные центральные проминирующие неправильной формы белесые помутнения в виде пятен. В отличие от зернистой дистрофии, нет четких зон между помутнениями роговицы. В развитой стадии вовлекаются эндотелий и десцеметова мембрана, на которой обычно имеются каплевидные наложения — гутты. Позднее из-за эндотелиальной декомпенсации строма отекает и утолщается. К 20-30 годам острота зрения обычно значительно снижена, как и чувствительность роговицы, появляется светобоязнь. Из-за рецидивирующих эрозий возникают приступы боли.
- Дистрофия роговицы Шнайдера — К1.
Дистрофия роговицы Шнайдера имеет аутосомнодоминантный тип наследования, ген UBIAD1 (UbiA prenyltransferase domain containing 1), локус 1p36. Морфологически в базальных клетках эпителия, слое Боумена, строме выявляются аномальные отложения внутри- и внеклеточных этерифицированных и неэтерифицированных фосфолипидов и холестерина. Диагноз ставится обычно во втором или третьем десятилетии при кристаллической форме, несколько позже — при беcкристаллической. Визуализируется центральное помутнение роговицы и/ или субэпителиальные кристаллы в виде снежинок. Затем появляется периферическое кольцевидное помутнение. Только 50% больных имеют кристаллы. Они иногда односторонни, редко регрессируют, могут появляться на поздних стадиях болезни. Острота зрения снижается с возрастом, появляются блики. При хорошем скотопическом зрении существенно страдает фотопическое. С возрастом уменьшается чувствительность роговицы. У родственников семьи, вовлеченной в патологический процесс, как с дистрофией роговицы, так и без нее возможна гиперлипопротеинемия.
- Врожденная стромальная дистрофия роговицы — К1.
- Крапчатая дистрофия роговицы — К1
Центральная дистрофия Франсуа-Нитенса (Francois, Neetens) Редкая форма дистрофии роговицы. Аутосомно-доминантный тип наследования. Вызывается мутациями гена PIP5K3. Вовлекается центр и периферия. Отмечаются маленькие овальные или неопределенной формы помутнения в передних слоях стромы с прозрачной окружающей тканью. Гистологически выявляются склеенные кератоциты, заполненные гликозаминогликанами. Острота зрения страдает редко. Болезнь не прогрессирует. В большинстве случаев протекает бессимптомно. Иногда отмечается легкая светобоязнь.
- Задняя аморфная дистрофия роговицы — К3.
- Центральная облаковидная дистрофия Франко — К4.
- Пре-десцеметова дистрофия роговицы — К4.
Эндотелиальные дистрофии
- Эндотелиальная дистрофия роговицы Фукса — К1, К2, К3.
Эндотелиальная дистрофия роговицы Фукса (ЭДРФ) — одна из часто встречающихся дистрофий. Наследование в большинстве случаев проследить не удается. При ранней форме выделен ген COL8A2, локус 1p34.3-p32, при поздней - найдена ассоциация с различными локусами — 13pter-q12.13 (ЭДРФ2), 18q21.2-q21.3 (ЭДРФ3), 20p13-p12 (ЭДРФ4), 5q33.1-q35.2 (ЭДРФ5), 10p11.2 (ЭДРФ6), 9p24.1-p22.1 (ЭДРФ7) и 15q25 (ЭДРФ8).
При световой микроскопии выявляется характерное утолщение десцеметовой мембраны. Патологически измененные эндотелиальные клетки вырабатывают гиалиновые наросты - гутты - на утолщенной десцеметовой мембране. Гутты погружаются в мембрану, сливаются между собой. Часть эндотелиальных клеток гибнет, оставшиеся увеличиваются по площади, теряют гексагональность. При отеке роговицы значительно нарушается ламеллярная архитектоника стромальных волокон. Случаи без явного наследования проявляются на пятом десятке лет. Варианты с ранним началом — 1-3 десятилетии.
Заболевание в процессе развития проходит стадии от «cornea guttata» до субэпителиального фиброза с васкуляризацией. На стадии «cornea guttata» при биомикроскопии на уровне десцеметовой мембраны просматриваются центральные изменения в виде «битой металлической крошки» или «шагреневой кожи» с или без «напыления» пигмента. Роговичных гутт при позднем варианте дистрофии больше, чем при раннем. При уменьшении числа эндотелиальных клеток менее чем 500 в мм2 эндотелиальный слой перестает выполнять свои основные барьерные и насосные функции — развивается отек стромы и эпителия. Появляются эпителиальные пузыри — буллы (буллезная стадия), разрыв которых ведет к появлению болезненных эрозий. Субэпителиальные фиброзные рубцы и периферическая поверхностная васкуляризация могут возникнуть на поздних стадиях хронического отека. Для пациентов характерно прогрессирующее снижение остроты зрения из-за эпителиального/стромального отека. Острота зрения может быть хуже утром из-за увеличения отека. Боль, светобоязнь и слезотечение возникают из-за крупных булл и эпителиальных эрозий.
- Задняя полиморфная дистрофия роговицы — К1 или К2.
При задней полиморфной дистрофии (ЗПД) тип наследования аутосомно-доминантный, выявлены различные локусы: для ЗПД1 — 20p11.2-q11.2; ЗПД2 — 1p34.3-p32.3; ЗПД3 — 10p11.22. Ген для ЗПД1 не выделен, для ЗПД2 — COL8A2, ЗПД3 — ZEB1. Морфологически на задней поверхности десцеметовой мембраны выявляются патологические слои коллагена с локальными веретенообразными или узелковыми разрастаниями. Среди эндотелия определяются участки многослойных эпителиоподобных клеток с микроворсинками и десмосомами. Заболевание проявляется часто асимметричными поражениями различной формы в глубоких слоях роговицы, включая узелковые, везикулезные (изолированные, в группах или сливные) и блистероподобные. Помутнения могут иметь вид «железнодорожных путей» и очаговых серых участков на уровне десцеметовой мембраны. Стромальный и эпителиальный отек из-за эндотелиальной декомпенсации развивается редко. Изменения в эндотелии часто не прогрессируют годами. Возможны медленное прогрессирование полиморфных везикул и утолщение десцеметовой мембраны на протяжении многих лет. Периферические иридокорнеальные сращения встречаются в 25% случаев. Примерно в 15% наблюдений уровень внутриглазного давления повышен. Эндотелиальные изменения часто протекают бессимптомно. Редко имеется значительное прогрессирующие снижение остроты зрения в результате стромальных помутнений.
- Врожденная эндотелиальная дистрофия — К1.
- X-связанная эндотелиальная дистрофия роговицы — К2.
Лечение
Эффективной патогенетической терапии на современном этапе развития медицины не существует. Симптоматическое местное лечение направлено на купирование нарушений передней поверхности роговицы и уменьшение ее отека при тех дистрофиях, где он имеет место. Для этого применяются любриканты, препараты, способствующие регенерации, мягкие контактные линзы, гипертонические растворы.
Перспективным представляется использование современных слезозаменителей. Хирургическое лечение может включать абразивные вмешательства на передней поверхности роговицы для улучшения адгезии эпителиального слоя к глубжележащим слоям и отчасти для уменьшения ее иррегулярности. ФТК является эффективным методом при удалении помутнений передних слоев роговой оболочки на фоне поверхностных и некоторых видов стромальных дистрофий. Кроме того, при выполнении ФТК удается устранить иррегулярный астигматизм и улучшить адгезию эпителия. При рецидивах заболевания процедуру можно повторять несколько раз.
При глубоких помутнениях, существенно снижающих остроту зрения, методом выбора является кератопластика. При относительно изолированном поражении слоев роговицы предпочтительными являются модификации послойной кератопластики (передняя послойная, глубокая послойная, эндотелиальная кератопластика). При одновременном вовлечении стромального и эндотелиального слоев целесообразно проводить сквозную кератопластику или ее модификации со сложным профилем операционного разреза. Как и после ФТК, после кератопластики вероятны рецидивы в сроки от 1 года до 20 лет, частота которых и степень снижения зрения зависят от вида дистрофии и могут варьировать индивидуально.
